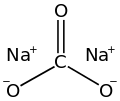
Sodium carbonate, Na2CO3·10H2O, (also known as washing soda, soda ash and soda crystals) is the inorganic compound with the formula Na2CO3 and its various hydrates. All forms are white, odourless, water-soluble salts that yield moderately alkaline solutions in water. Historically, it was extracted from the ashes of plants growing in sodium-rich soils. Because the ashes of these sodium-rich plants were noticeably different from ashes of wood (once used to produce potash), sodium carbonate became known as "soda ash".[13] It is produced in large quantities from sodium chloride and limestone by the Solvay process.
https://en.wikipedia.org/wiki/Sodium_carbonate
Sodium ferrocyanide is the sodium salt of the coordination compound of formula [Fe(CN)6]4−. In its hydrousform, Na4Fe(CN)6 • 10H2O (sodium ferrocyanide decahydrate), it is sometimes known as yellow prussiate of soda. It is a yellow crystalline solid that is soluble in water and insoluble in alcohol. The yellow color is the color of ferrocyanide anion. Despite the presence of the cyanide ligands, sodium ferrocyanide has low toxicity(acceptable daily intake 0–0.025 mg/kg body weight[2]). The ferrocyanides are less toxic than many salts of cyanide, because they tend not to release free cyanide.[3] However, like all ferrocyanide salt solutions, addition of an acid can result in the production of hydrogen cyanide gas, which is toxic.

Uses[edit]
When combined with iron, it converts to a deep blue pigment called Prussian blue, FeIII
4[FeII
(CN)
6]
3.[4] It is used as a stabilizer for the coating on welding rods. In the petroleum industry, it is used for removal of mercaptans.
In the EU, ferrocyanides (E 535–538) were, as of 2018, solely authorized as additives in salt and salt substitutes, where they serve as an anticaking agents. Kidneys are the organ for ferrocyanide toxicity, but ferrocyanides are of no safety concern at the levels at which they are used.[5]
Production[edit]
Sodium ferrocyanide is produced industrially from hydrogen cyanide, ferrous chloride, and calcium hydroxide, the combination of which affords Ca2[Fe(CN)6] • 11H2O. A solution of this salt is then treated with sodium salts to precipitate the mixed calcium-sodium salt CaNa2[Fe(CN)6], which in turn is treated with sodium carbonate to give the tetrasodium salt.[6]
https://en.wikipedia.org/wiki/Sodium_ferrocyanide
Prussian blue (also known as Berlin blue or, in painting, Parisian or Paris blue) is a dark blue pigmentproduced by oxidation of ferrous ferrocyanide salts. It has the chemical formula FeIII
4[FeII
(CN)
6]
3. Turnbull's blue is chemically identical, but is made from different reagents, and its slightly different color stems from different impurities.
Prussian blue was the first modern synthetic pigment. It is prepared as a very fine colloidal dispersion, because the compound is not soluble in water. It contains variable amounts[1] of other ions and its appearance depends sensitively on the size of the colloidal particles. The pigment is used in paints, and it is the traditional "blue" in blueprints and aizuri-e (藍摺り絵) Japanese woodblock prints.
In medicine, orally administered Prussian blue is used as an antidote for certain kinds of heavy metal poisoning, e.g., by thallium(I) and radioactive isotopes of caesium. The therapy exploits the compound's ion-exchange properties and high affinity for certain "soft" metal cations.
It is on the World Health Organization's List of Essential Medicines, the most important medications needed in a basic health system.[2] Prussian blue lent its name to prussic acid (hydrogen cyanide) derived from it. In German, hydrogen cyanide is called Blausäure ("blue acid"). French chemist Joseph Louis Gay-Lussac gave cyanide its name, from the Ancient Greek word κύανος (kyanos, "blue"), because of its Prussian blue color.
https://en.wikipedia.org/wiki/Prussian_blue
Iron(III) sulfate (or ferric sulfate), is a family of inorganic compounds with the formula Fe2(SO4)3(H2O)n. A variety of hydrates are known, in fact are the most commonly encountered form of "ferric sulfate". Solutions are used in dyeing as a mordant, and as a coagulant for industrial wastes. It is also used in pickling baths for aluminum and steel.[2][3]
Generally, ferric sulfate is used as a solution generated from iron wastes. The actual speciation is vague but its applications do not demand high purity materials. As such iron(III) sulfate is generated and handled as an aqueous solution. It is produced on a large scale by treating sulfuric acid, a hot solution of ferrous sulfate, and an oxidizing agent. Typical oxidizing agents include chlorine, nitric acid, and hydrogen peroxide.[4]
- 2 FeSO4 + H2SO4 + H2O2 → Fe2(SO4)3 + 2 H2O
https://en.wikipedia.org/wiki/Iron(III)_sulfate
Potash (/ˈpɒtæʃ/) includes various mined and manufactured salts that contain potassium in water-soluble form.[1]The name derives from pot ash, which refers to plant ashes or wood ash soaked in water in a pot, which was the primary means of manufacturing the product before the Industrial Era. The word "potassium" is derived from "potash".[2]
Potash is produced worldwide in amounts exceeding 90 million tonnes (40 million tonnes K2O equivalent) per year, mostly for use in fertilizer. Various kinds of fertilizer-potash constitute the single greatest industrial use of the element potassium in the world. Potassium was first derived in 1807 by electrolysis of caustic potash (potassium hydroxide).[3]
Terminology[edit]
Potash refers to potassium compounds and potassium-bearing materials, most commonly potassium carbonate. The word "potash" originates from the Middle Dutch "potaschen", denoting "pot ashes" in 1477. [4] The old method of making potassium carbonate (K
2CO
3) was by collecting or producing wood ash (the occupation of ash burners), leaching the ashes, and then evaporating the resulting solution in large iron pots, which left a white residue denominated "pot ash".[5] Approximately 10% by weight of common wood ash can be recovered as potash.[6][7] Later, "potash" became widely applied to naturally occurring potassium salts and the commercial product derived from them,[8] although it most probably derived its name (where it was used) from the anion of the acid that replaced the carbonate moiety, a common equivocative use of "potash" for "potassium".
The following table lists a number of potassium compounds which have "potash" in their traditional names:
| Common name | Chemical name (Formula) | |
|---|---|---|
| Potash fertilizer | c.1942 potassium carbonate (K2CO3); c.1950 any one or more of potassium chloride (KCl), potassium sulfate (K2SO4) or potassium nitrate (KNO3).[9][10] Does not contain potassium oxide (K2O), which plants do not take up.[11] However, the amount of potassium is often reported as K2O equivalent (that is, how much it would be if in K2O form), to allow apples-to-apples comparison between different fertilizers using different types of potash. | |
| Caustic potash or potash lye | potassium hydroxide (KOH) | |
| Carbonate of potash,salts of tartar, or pearl ash | potassium carbonate (K2CO3) | |
| Chlorate of potash | potassium chlorate (KClO3) | |
| Muriate of potash (MOP) | potassium chloride (KCl:NaCl = 95:5 or higher)[1] | |
| Nitrate of potash or saltpeter | potassium nitrate (KNO3) | |
| Sulfate of potash (SOP) | potassium sulfate (K2SO4) | |
| Permanganate of potash | potassium permanganate (KMnO4) | |
Production[edit]
All commercial potash deposits come originally from evaporite deposits and are often buried deep below the earth's surface. Potash ores are typically rich in potassium chloride (KCl), sodium chloride (NaCl) and other salts and clays, and are typically obtained by conventional shaft mining with the extracted ore ground into a powder.[12] Other methods include dissolution mining and evaporation methods from brines.
In the evaporation method, hot water is injected into the potash which is dissolved and then pumped to the surface where it is concentrated by solar induced evaporation. Amine reagents are then added to either the mined or evaporated solutions. The amine coats the KCl but not NaCl. Air bubbles cling to the amine + KCl and float it to the surface while the NaCl and clay sink to the bottom. The surface is skimmed for the amine + KCl which is then dried and packaged for use as a K rich fertilizer—KCl dissolves readily in water and is available quickly for plant nutrition.[13]
Potash deposits can be found all over the world. At present, deposits are being mined in Canada, Russia, China, Belarus, Israel, Germany, Chile, the United States, Jordan, Spain, the United Kingdom, Uzbekistan and Brazil,[14] with the most significant deposits present in Saskatchewan, Canada.[7]
Occupational hazards[edit]
Excessive respiratory disease has been a concern for potash miners throughout history due to environmental hazards, such as radon and asbestos. Potash miners are liable to develop silicosis. Based on a study done between 1977 and 1987, cardiovascular disease among potash workers, the overall mortality rates were low, but a noticeable difference in above-ground workers was documented.[15]
History of production[edit]


Potash (especially potassium carbonate) has been used in bleaching textiles, making glass, ceramic, and making soap, since the Bronze Age.[citation needed] Potash was principally obtained by leaching the ashes of land and sea plants.
Beginning in the 14th century potash was mined in Ethiopia. One of the world's largest deposits, 140 to 150 million tons, is located in the Dallol area of the Afar Region.[16]
Potash was one of the most important industrial chemicals. It was refined from the ashes of broadleaved trees and produced primarily in the forested areas of Europe, Russia, and North America. Although methods for producing artificial alkalis were invented in the late eighteenth century, these did not become economical until the late nineteenth century and so the dependence on organic sources of potash remained.
Potash became an important international trade commodity in Europe from at least the early fourteenth century. It is estimated that European imports of potash required 6 or more million cubic metres each year from the early seventeenth century.[17]
Between 1420 and 1620, the primary exporting cities for wood-derived potash were Danzig, Königsberg and Riga. From the 1640s, geopolitical disruptions meant that the centres of export moved from the Baltic to Archangel, Russia. In 1700, Russian ash was dominant though Danzig remained notable for the quality of its potash. In the late fifteenth century, London was the lead importer due to its position as the centre of soft soap making while the Dutch dominated as suppliers and consumers in the sixteenth century.[18]
The first U.S. patent of any kind was issued in 1790 to Samuel Hopkins for an improvement "in the making of Pot ash and Pearl ash by a new Apparatus and Process".[19] Pearl ash was a purer quality made by calcination of potash in a reverberatory furnace or kiln. Potash pits were once used in England to produce potash that was used in making soap for the preparation of wool for yarn production.
See also[edit]
https://en.wikipedia.org/wiki/Potash
An evaporite (/ɪˈvæpəˌraɪt/) is a water-soluble sedimentary mineral deposit that results from concentration and crystallization by evaporation from an aqueous solution.[1] There are two types of evaporite deposits: marine, which can also be described as ocean deposits, and non-marine, which are found in standing bodies of water such as lakes. Evaporites are considered sedimentary rocks and are formed by chemical sediments.

Formation of evaporite rocks[edit]
Although all water bodies on the surface and in aquifers contain dissolved salts, the water must evaporate into the atmosphere for the minerals to precipitate. For this to happen, the water body must enter a restricted environment where water input into this environment remains below the net rate of evaporation. This is usually an arid environment with a small basin fed by a limited input of water. When evaporation occurs, the remaining water is enriched in salts, and they precipitate when the water becomes supersaturated.
Evaporite depositional environments[edit]
Marine evaporites[edit]

Marine evaporites tend to have thicker deposits and are usually the focus of more extensive research.[2] They also have a system of evaporation. When scientists evaporate ocean water in a laboratory, the minerals are deposited in a defined order that was first demonstrated by Usiglio in 1884.[2] The first phase of the experiment begins when about 50% of the original water depth remains. At this point, minor carbonates begin to form.[2] The next phase in the sequence comes when the experiment is left with about 20% of its original level. At this point, the mineral gypsum begins to form, which is then followed by halite at 10%,[2] excluding carbonate minerals that tend not to be evaporites. The most common minerals that are generally considered to be the most representative of marine evaporites are calcite, gypsum and anhydrite, halite, sylvite, carnallite, langbeinite, polyhalite, and kainite. Kieserite(MgSO4) may also be included, which often will make up less than four percent of the overall content.[2] However, there are approximately 80 different minerals that have been reported found in evaporite deposits (Stewart, 1963; Warren, 1999), though only about a dozen are common enough to be considered important rock formers.[2]
Non-marine evaporites[edit]
Non-marine evaporites are usually composed of minerals that are not common in marine environments because in general the water from which non-marine evaporite precipitates has proportions of chemical elements different from those found in the marine environments.[2] Common minerals that are found in these deposits include blödite, borax, epsomite, gaylussite, glauberite, mirabilite, thenardite and trona. Non-marine deposits may also contain halite, gypsum, and anhydrite, and may in some cases even be dominated by these minerals, although they did not come from ocean deposits. This, however, does not make non-marine deposits any less important; these deposits often help to paint a picture into past Earth climates. Some particular deposits even show important tectonic and climatic changes. These deposits also may contain important minerals that help in today's economy.[3] Thick non-marine deposits that accumulate tend to form where evaporation rates will exceed the inflow rate, and where there is sufficient soluble supplies. The inflow also has to occur in a closed basin, or one with restricted outflow, so that the sediment has time to pool and form in a lake or other standing body of water.[3] Primary examples of this are called "saline lake deposits".[3] Saline lakes includes things such as perennial lakes, which are lakes that are there year-round, playa lakes, which are lakes that appear only during certain seasons, or any other terms that are used to define places that hold standing bodies of water intermittently or year-round. Examples of modern non-marine depositional environments include the Great Salt Lake in Utah and the Dead Sea, which lies between Jordan and Israel.
Evaporite depositional environments that meet the above conditions include:
- Graben areas and half-grabens within continental rift environments fed by limited riverine drainage, usually in subtropical or tropical environments
- Example environments at the present that match this is the Denakil Depression, Ethiopia; Death Valley, California
- Graben environments in oceanic rift environments fed by limited oceanic input, leading to eventual isolation and evaporation
- Examples include the Red Sea, and the Dead Sea in Jordan and Israel
- Internal drainage basins in arid to semi-arid temperate to tropical environments fed by ephemeral drainage
- Example environments at the present include the Simpson Desert, Western Australia, the Great Salt Lake in Utah
- Non-basin areas fed exclusively by groundwater seepage from artesian waters
- Example environments include the seep-mounds of the Victoria Desert, fed by the Great Artesian Basin, Australia
- Restricted coastal plains in regressive sea environments
- Examples include the sabkha deposits of Iran, Saudi Arabia, and the Red Sea; the Garabogazköl of the Caspian Sea
- Drainage basins feeding into extremely arid environments
The most significant known evaporite depositions happened during the Messinian salinity crisis in the basin of the Mediterranean.
Evaporitic formations[edit]

Evaporite formations need not be composed entirely of halite salt. In fact, most evaporite formations do not contain more than a few percent of evaporite minerals, the remainder being composed of the more typical detrital clasticrocks and carbonates. Examples of evaporite formations include occurrences of evaporite sulfur in Eastern Europe and West Asia.[4]
For a formation to be recognised as evaporitic it may simply require recognition of halite pseudomorphs, sequences composed of some proportion of evaporite minerals, and recognition of mud crack textures or other textures.
Economic importance of evaporites[edit]
Evaporites are important economically because of their mineralogy, their physical properties in-situ, and their behaviour within the subsurface.
Evaporite minerals, especially nitrate minerals, are economically important in Peru and Chile. Nitrate minerals are often mined for use in the production on fertilizer and explosives.
Thick halite deposits are expected to become an important location for the disposal of nuclear waste because of their geologic stability, predictable engineering and physical behaviour, and imperviousness to groundwater.
Halite formations are famous for their ability to form diapirs, which produce ideal locations for trapping petroleum deposits.
Halite deposits are often mined for use as salt.
Major groups of evaporite minerals[edit]

This is a chart that shows minerals that form the marine evaporite rocks, they are usually the most common minerals that appear in this kind of deposit.
Mineral Class Mineral name Chemical Composition Chlorides Halite NaCl Sylvite KCl Carnallite KMgCl3 · 6 H2O Kainite KMg(SO4)Cl · 3 H2O Sulfates Anhydrite CaSO4 Gypsum CaSO4 · 2 H2O Kieserite MgSO4 · H2O Langbeinite K2Mg2(SO4)3 Polyhalite K2Ca2Mg(SO4)6 · H2O Carbonates Dolomite CaMg(CO3)2 Calcite CaCO3 Magnesite MgCO3

- Halides: halite, sylvite (KCl), and fluorite
- Sulfates: such as gypsum, barite, and anhydrite
- Nitrates: nitratine (soda niter) and niter
- Borates: typically found in arid-salt-lake deposits plentiful in the southwestern US. A common borate is borax, which has been used in soaps as a surfactant.
- Carbonates: such as trona, formed in inland brine lakes.
- Some evaporite minerals, such as Hanksite, are from multiple groups.
Evaporite minerals start to precipitate when their concentration in water reaches such a level that they can no longer exist as solutes.
The minerals precipitate out of solution in the reverse order of their solubilities, such that the order of precipitation from sea water is:
- Calcite (CaCO3) and dolomite (CaMg(CO3)2)
- Gypsum (CaSO4 • 2H2O) and anhydrite (CaSO4).
- Halite (i.e. common salt, NaCl)
- Potassium and magnesium salts
The abundance of rocks formed by seawater precipitation is in the same order as the precipitation given above. Thus, limestone (calcite) and dolomite are more common than gypsum, which is more common than halite, which is more common than potassium and magnesium salts.
Evaporites can also be easily recrystallized in laboratories in order to investigate the conditions and characteristics of their formation.
Possible evaporites on Titan[edit]
Recent evidence from satellite observations[5] and laboratory experiments[6] suggest evaporites are likely present on the surface of Titan, Saturn's largest moon. Instead of water oceans, Titan hosts lakes and seas of liquid hydrocarbons (mainly methane) with many soluble hydrocarbons, such as acetylene,[7]that can evaporate out of solution. Evaporite deposits cover large regions of Titan's surface, mainly along the coastlines of lakes or in isolated basins(Lacunae) that are equivalent to salt pans on Earth.[8]
See also[edit]
| Wikimedia Commons has media related to Evaporite. |
https://en.wikipedia.org/wiki/Evaporite
Thallium(I)[edit]
The thallium(I) halides are stable. In keeping with the large size of the Tl+ cation, the chloride and bromide have the caesium chloride structure, while the fluoride and iodide have distorted sodium chloride structures. Like the analogous silver compounds, TlCl, TlBr, and TlI are photosensitive and display poor solubility in water.[17] The stability of thallium(I) compounds demonstrates its differences from the rest of the group: a stable oxide, hydroxide, and carbonateare known, as are many chalcogenides.[18]
The double salt Tl
4(OH)
2CO
3 has been shown to have hydroxyl-centred triangles of thallium, [Tl
3(OH)]2+
, as a recurring motif throughout its solid structure.[19]
The metalorganic compound thallium ethoxide (TlOEt, TlOC2H5) is a heavy liquid (ρ 3.49 g·cm−3, m.p. −3 °C),[20] often used as a basic and soluble thallium source in organic and organometallic chemistry.[21]
Organothallium compounds[edit]
Organothallium compounds tend to be thermally unstable, in concordance with the trend of decreasing thermal stability down group 13. The chemical reactivity of the Tl–C bond is also the lowest in the group, especially for ionic compounds of the type R2TlX. Thallium forms the stable [Tl(CH3)2]+ ion in aqueous solution; like the isoelectronic Hg(CH3)2 and [Pb(CH3)2]2+, it is linear. Trimethylthallium and triethylthallium are, like the corresponding gallium and indium compounds, flammable liquids with low melting points. Like indium, thallium cyclopentadienyl compounds contain thallium(I), in contrast to gallium(III).[22]
History[edit]
Thallium (Greek θαλλός, thallos, meaning "a green shoot or twig")[23] was discovered by William Crookes and Claude Auguste Lamy, working independently, both using flame spectroscopy (Crookes was first to publish his findings, on March 30, 1861).[24] The name comes from thallium's bright green spectral emission lines.[25]
After the publication of the improved method of flame spectroscopy by Robert Bunsen and Gustav Kirchhoff[26] and the discovery of caesium and rubidiumin the years 1859 to 1860, flame spectroscopy became an approved method to determine the composition of minerals and chemical products. Crookes and Lamy both started to use the new method. Crookes used it to make spectroscopic determinations for tellurium on selenium compounds deposited in the lead chamber of a sulfuric acid production plant near Tilkerode in the Harz mountains. He had obtained the samples for his research on selenium cyanide from August Hofmann years earlier.[27][28] By 1862, Crookes was able to isolate small quantities of the new element and determine the properties of a few compounds.[29] Claude-Auguste Lamy used a spectrometer that was similar to Crookes' to determine the composition of a selenium-containing substance which was deposited during the production of sulfuric acid from pyrite. He also noticed the new green line in the spectra and concluded that a new element was present. Lamy had received this material from the sulfuric acid plant of his friend Fréd Kuhlmann and this by-product was available in large quantities. Lamy started to isolate the new element from that source.[30] The fact that Lamy was able to work ample quantities of thallium enabled him to determine the properties of several compounds and in addition he prepared a small ingot of metallic thallium which he prepared by remelting thallium he had obtained by electrolysis of thallium salts.[citation needed]
As both scientists discovered thallium independently and a large part of the work, especially the isolation of the metallic thallium was done by Lamy, Crookes tried to secure his own priority on the work. Lamy was awarded a medal at the International Exhibition in London 1862: For the discovery of a new and abundant source of thallium and after heavy protest Crookes also received a medal: thallium, for the discovery of the new element. The controversy between both scientists continued through 1862 and 1863. Most of the discussion ended after Crookes was elected Fellow of the Royal Society in June 1863.[31][32]
The dominant use of thallium was the use as poison for rodents. After several accidents the use as poison was banned in the United States by Presidential Executive Order 11643 in February 1972. In subsequent years several other countries also banned its use.[33]
https://en.wikipedia.org/wiki/Thallium#Thallium(I)
Hard and soft classification[edit]
Lewis acids and bases are commonly classified according to their hardness or softness. In this context hard implies small and nonpolarizable and soft indicates larger atoms that are more polarizable.
- typical hard acids: H+, alkali/alkaline earth metal cations, boranes, Zn2+
- typical soft acids: Ag+, Mo(0), Ni(0), Pt2+
- typical hard bases: ammonia and amines, water, carboxylates, fluoride and chloride
- typical soft bases: organophosphines, thioethers, carbon monoxide, iodide
For example, an amine will displace phosphine from the adduct with the acid BF3. In the same way, bases could be classified. For example, bases donating a lone pair from an oxygen atom are harder than bases donating through a nitrogen atom. Although the classification was never quantified it proved to be very useful in predicting the strength of adduct formation, using the key concepts that hard acid—hard base and soft acid—soft base interactions are stronger than hard acid—soft base or soft acid—hard base interactions. Later investigation of the thermodynamics of the interaction suggested that hard—hard interactions are enthalpy favored, whereas soft—soft are entropy favored.
Quantifying Lewis acidity[edit]
Many methods have been devised to evaluate and predict Lewis acidity. Many are based on spectroscopic signatures such as shifts NMR signals or IR bands e.g. the Gutmann-Beckett method and the Childs[13] method.
The ECW model is a quantitative model that describes and predicts the strength of Lewis acid base interactions, −ΔH. The model assigned E and C parameters to many Lewis acids and bases. Each acid is characterized by an EA and a CA. Each base is likewise characterized by its own EB and CB. The E and C parameters refer, respectively, to the electrostatic and covalent contributions to the strength of the bonds that the acid and base will form. The equation is
- −ΔH = EAEB + CACB + W
The W term represents a constant energy contribution for acid–base reaction such as the cleavage of a dimeric acid or base. The equation predicts reversal of acids and base strengths. The graphical presentations of the equation show that there is no single order of Lewis base strengths or Lewis acid strengths.[14] [15] and that single property scales are limited to a smaller range of acids or bases.
https://en.wikipedia.org/wiki/Lewis_acids_and_bases#Hard_and_soft_classification
A colloid is a mixture in which one substance of microscopically dispersed insoluble particles are suspended throughout another substance. However, some definitions specify that the particles must be dispersed in a liquid,[1] and others extend the definition to include substances like aerosols and gels. The term colloidal suspension refers unambiguously to the overall mixture (although a narrower sense of the word suspension is distinguished from colloids by larger particle size). A colloid has a dispersed phase (the suspended particles) and a continuous phase (the medium of suspension). The dispersed phase particles have a diameter of approximately 1 nanometre to 1 micrometre.[2][3]
Some colloids are translucent because of the Tyndall effect, which is the scattering of light by particles in the colloid. Other colloids may be opaque or have a slight color.
Colloidal suspensions are the subject of interface and colloid science. This field of study was introduced in 1845 by Italian chemist Francesco Selmi[4] and further investigated since 1861 by Scottish scientist Thomas Graham.[5]
https://en.wikipedia.org/wiki/Colloid
Iron(II) chloride, also known as ferrous chloride, is the chemical compound of formula FeCl2. It is a paramagnetic solid with a high melting point. The compound is white, but typical samples are often off-white. FeCl2 crystallizes from water as the greenish tetrahydrate, which is the form that is most commonly encountered in commerce and the laboratory. There is also a dihydrate. The compound is highly soluble in water, giving pale green solutions.

https://en.wikipedia.org/wiki/Iron(II)_chloride
Calcium hydroxide (traditionally called slaked lime) is an inorganic compound with the chemical formula Ca(OH)2. It is a colorless crystal or white powder and is produced when quicklime (calcium oxide) is mixed or slaked with water. It has many names including hydrated lime, caustic lime, builders' lime, slack lime, cal, and pickling lime. Calcium hydroxide is used in many applications, including food preparation, where it has been identified as E number E526. Limewater is the common name for a saturated solution of calcium hydroxide.

Properties[edit]
Calcium hydroxide is poorly soluble in water with a retrograde solubility increasing from 0.66 g/L at 100 °C to 1.89 g/L at 0 °C. With a solubility product Ksp of 5.5×10−6 at T = ?[clarification needed].[citation needed] its dissociation in water is large enough that its solutions are basic according to the following dissolution reaction:
- Ca(OH)2 → Ca2+ + 2 OH−
At ambient temperature, calcium hydroxide (portlandite) dissolves in pure water to produce an alkaline solution with a pH of about 12.5. Calcium hydroxide solutions can cause chemical burns. At high pH value due to a common-ion effect with the hydroxide anion OH−
, its solubility drastically decreases. This behavior is relevant to cement pastes. Aqueous solutions of calcium hydroxide are called limewater and are medium-strength bases, which reacts with acids and can attack some metals such as aluminium (amphoteric hydroxide dissolving at high pH), while protecting other metals, such as iron and steel, from corrosion by passivation of their surface. Limewater turns milky in the presence of carbon dioxide due to formation of calcium carbonate, a process called carbonatation:
- Ca(OH)2 + CO2 → CaCO3 + H2O
When heated to 512 °C, the partial pressure of water in equilibrium with calcium hydroxide reaches 101 kPa (normal atmospheric pressure), which decomposes calcium hydroxide into calcium oxide and water:[8]
- Ca(OH)2 → CaO + H2O
Structure, preparation, occurrence[edit]

Calcium hydroxide adopts a polymeric structure, as do all metal hydroxides. The structure is identical to that of Mg(OH)2 (brucite structure); i.e., the cadmium iodide motif. Strong hydrogen bonds exist between the layers.[9]
Calcium hydroxide is produced commercially by treating lime with water:
- CaO + H2O → Ca(OH)2
In the laboratory it can be prepared by mixing aqueous solutions of calcium chloride and sodium hydroxide. The mineral form, portlandite, is relatively rare but can be found in some volcanic, plutonic, and metamorphic rocks. It has also been known to arise in burning coal dumps.
The positively charged ionized species CaOH+ has been detected in the atmosphere of S-type stars.[10]
Uses[edit]
Calcium hydroxide is commonly used to prepare lime mortar.
One significant application of calcium hydroxide is as a flocculant, in water and sewage treatment. It forms a fluffy charged solid that aids in the removal of smaller particles from water, resulting in a clearer product. This application is enabled by the low cost and low toxicity of calcium hydroxide. It is also used in fresh-water treatment for raising the pH of the water so that pipes will not corrode where the base water is acidic, because it is self-regulating and does not raise the pH too much.
It is also used in the preparation of ammonia gas (NH3), using the following reaction:
Another large application is in the paper industry, where it is an intermediate in the reaction in the production of sodium hydroxide. This conversion is part of the causticizing step in the Kraft process for making pulp.[9] In the causticizing operation, burned lime is added to green liquor, which is a solution primarily of sodium carbonateand sodium sulfate produced by dissolving smelt, which is the molten form of these chemicals from the recovery furnace.
Food industry[edit]
Because of its low toxicity and the mildness of its basic properties, slaked lime is widely used in the food industry:
- In USDA certified food production in plants and livestock[19]
- To clarify raw juice from sugarcane or sugar beets in the sugar industry, (see carbonatation)
- To process water for alcoholic beverages and soft drinks
- Pickle cucumbers and other foods
- To make Chinese century eggs
- In maize preparation: removes the cellulose hull of maize kernels (see nixtamalization)
- To clear a brine of carbonates of calcium and magnesium in the manufacture of salt for food and pharmaceutical uses
- In fortifying (Ca supplement) fruit drinks, such as orange juice, and infant formula
- As a digestive aid (called Choona, used in India in paan, a mixture of areca nuts, calcium hydroxide and a variety of seeds wrapped in betel leaves)
- As a substitute for baking soda in making papadam
- In the removal of carbon dioxide from controlled atmosphere produce storage rooms
- In the preparation of mushroom growing substrates[20]
Native American uses[edit]

In Spanish, calcium hydroxide is called cal. Maize cooked with cal (in a process of nixtamalization) becomes hominy (nixtamal), which significantly increases the bioavailability of niacin (vitamin B3), and is also considered tastier and easier to digest.
In chewing coca leaves, calcium hydroxide is usually chewed alongside to keep the alkaloid stimulants chemically available for absorption by the body. Similarly, Native Americans traditionally chewed tobacco leaves with calcium hydroxide derived from burnt mollusc shells to enhance the effects. It has also been used by some indigenous American tribes as an ingredient in yopo, a psychedelic snuff prepared from the beans of some Anadenanthera species.[21]
Asian uses[edit]
Calcium hydroxide is typically added to a bundle of areca nut and betel leaf called "paan" to keep the alkaloidstimulants chemically available to enter the bloodstream via sublingual absorption.
It is used in making naswar (also known as nass or niswar), a type of dipping tobacco made from fresh tobacco leaves, calcium hydroxide (chuna or soon), and wood ash. It is consumed most in the Pathan diaspora, Afghanistan, Pakistan, India and Bangladesh. Villagers also use calcium hydroxide to paint their mud houses in Afghanistan, Pakistan and India.
Health risks[edit]
Unprotected exposure to Ca(OH)2 can cause severe skin irritation, chemical burns, blindness, lung damage or rashes.[6]
See also[edit]
- Baralyme (carbon dioxide absorbent)
- Cement
- Lime mortar
- Lime plaster
- Plaster
- Magnesium hydroxide (less alkaline due to a lower solubility product)
- Soda lime (carbon dioxide absorbent)
- Whitewash
https://en.wikipedia.org/wiki/Calcium_hydroxide
Whitewash, or calcimine, kalsomine, calsomine, or lime paint is a type of paint made from slaked lime (calcium hydroxide, Ca(OH)2) or chalk calcium carbonate, (CaCO3), sometimes known as "whiting". Various other additives are sometimes used.
Whitewash cures through a reaction with carbon dioxide in the atmosphere to form calcium carbonate in the form of calcite, a type of reaction generally known as carbonation or by the more specific term, carbonatation.
It is usually applied to exteriors; however, it has been traditionally used in interiors of food preparation areas, particularly rural dairies, because of its mildly antibacterial properties. Whitewash can be tinted for decorative use and is sometimes painted inside structures such as the hallways of apartment buildings, however it can rub off onto clothing to a small degree. In Britain and Ireland, whitewash was used historically in interiors and exteriors of workers' cottages and still retains something of this association with rural poverty. In the United States, a similar attitude is expressed in the old saying: "Too proud to whitewash and too poor to paint."[1]
Whitewash is especially compatible with masonry because it is absorbed easily and the resultant chemical reaction hardens the medium.
Lime wash is pure slaked lime in water. It produces a unique surface glow due to the double refraction of calcite crystals. Limewash and whitewash both cure to become the same material.
When whitewash or limewash is initially applied, it has very low opacity, which can lead novices to overthicken the paint. Drying increases opacity and subsequent curing increases opacity even further.
Limewash relies on being drawn into a substrate unlike a modern paint that adheres to the surface. The process of being drawn in needs to be controlled by damping down. If a wall is not damped, it can leave the lime and pigments on the surface powdery; if the wall is saturated, then there is no surface tension and this can result in failure of the limewash. Damping down is not difficult but it does need to be considered before application of the limewash.[2]
Additives[edit]
Additives traditionally used include water glass, glue, egg white, Portland cement, salt, soap, milk, flour, and soil.
Whitewash is sometimes coloured with earths to achieve colours spanning the range of broken white, cream, yellow and a range of browns.
The blue laundry dye (such as Reckett's "Dolly Blue" in the UK, Ireland and Australia, Loulaki in Greece, or Mrs. Stewart's Bluing in North America) formerly widely used to give a bright tinge to boiled white textiles was a common 19th century addition .
Historically, pig's blood was added to give the colour Suffolk pink, a colour still widely used on house exteriors in some areas of the UK. If animal blood is applied excessively, however, its iron oxide can compromise the lime binder's strength.
Pozzolanic materials are occasionally added to give a much harder wearing paint finish. This addition, however, creates a short open time and therefore requires timely application of the altered paint.
Linseed oil is sometimes added (typically 0.5-2%) to improve adhesion on difficult surfaces.
Cement addition makes a harder wearing paint in white or grey. Open time is short, so this is added at point of use. However, the use of cement restricts the breathable aspects of the limewash and is advised to not be applied to historic buildings.
Dilute glues improve paint toughness.
Wheat flour has been used as a strength enhancing binder. Salt is often added to prevent mold.
Limitations[edit]
Basic limewash can be inadequate in its ability to prevent rain-driven water ingress. Additives are being developed but these have the potential for affecting free vapor permeability. For this reason silicate paints, more common in Germany, are gaining popularity in the UK over limewash.
https://en.wikipedia.org/wiki/Whitewash
Baralyme is a mixture of 80% calcium hydroxide and 20% barium hydroxide compounds[1][2][3] that is used as an alternative to soda lime to absorb the exhaled carbon dioxide in a closed circuit anesthetic system.[4]
The substance has been used for carbon dioxide scrubbing in diving bells and the U.S Navy's engineered SEALAB's I, II, and the failed SEALAB III.[citation needed]
https://en.wikipedia.org/wiki/Baralyme
Soda lime is a mixture of NaOH & CaO chemicals, used in granular form in closed breathing environments, such as general anaesthesia, submarines, rebreathers and recompression chambers, to remove carbon dioxide from breathing gases to prevent CO2 retention and carbon dioxide poisoning.[1][2]
It is made by treating slaked lime with concentrated sodium hydroxide solution.
Chemical components[edit]
The main components of soda lime are
- Calcium oxide, CaO (about 75%)
- Water, H2O (about 20%)
- Sodium hydroxide, NaOH (about 3%)
- Potassium hydroxide, KOH (about 1%).
Anaesthetic use[edit]
During the administration of general anaesthesia, the gases expired by a patient, which contain carbon dioxide, are passed through an anaesthetic machinebreathing circuit filled with soda lime granules.[1] Medical-grade soda lime includes an indicating dye that changes color when the soda lime reaches its carbon dioxide absorbing capacity.
To ensure that a soda lime canister (CO2 absorber) is functioning properly, it should not be used if the indicating dye is activated. Standard anaesthesia machines typically contain up to 2 kg of soda lime granules.
Lithium hydroxide (LiOH) is the alkali hydroxide with the lowest molecular weight (Na: 23 g/mol; Li: 7 g/mol) and is therefore used as CO2 absorbent in space flights since the Apollo program to spare weight at launch. During Apollo 13 flight, the crew sheltered in the lunar module started suffering from high CO2 levels and had to adapt spare absorbent cartridges from the Apollo capsule to the LEM system.
Recent generation of CO2 absorbents have been developed to reduce the risk of formation of toxic by-products as a result of the interaction between the absorbent and inhaled anesthetics (halothane). Some absorbents made from lithium hydroxide (LiOH) are also available for this purpose.
Rebreather use[edit]
Exhaled gas must be passed through a carbon dioxide scrubber where the carbon dioxide is absorbed before the gas is made available to be breathed again. In rebreathers the scrubber is a part of the breathing loop.[2][3] Color indicating dye was removed from US Navy fleet use in 1996 when it was suspected of releasing chemicals into the circuit.[4] In larger environments, such as recompression chambers or submarines, a fan is used to maintain the flow of gas through the scrubbing canister.[2]
Chemical reaction[edit]
The overall reaction is:
- CO2 + CaO→ CaCO3 + heat (in the presence of water)
Each mole of CO2 (44 g) reacting with calcium hydroxide produces one mole of water (18 g).
The reaction can be considered as a strong-base-catalysed, water-facilitated reaction.[5]
The reaction mechanism of carbon dioxide with soda lime can be decomposed in three elementary steps:
- 1) CO2(g) → CO2(aq) (CO2 dissolves in water – slow and rate-determining)
- 2) CO2(aq) + NaOH → NaHCO3 (bicarbonate formation at high pH)
- 3) NaHCO3 + CaO→ CaCO3 + NaOH (NaOH recycled to step 2 – hence a catalyst)
This sequence of reactions explains the catalytic role played by sodium hydroxide in the system and why soda lime is faster in chemical reactivity than calcium hydroxide alone.[6] The moist NaOH impregnates the surface and the porosity of calcium hydroxide grains with a high specific surface area.[7] It reacts much more quickly and so contributes to a faster elimination of the CO2 from the rebreathing circuit. The formation of water by the reaction and the moisture from the respiration also act as a solvent for the reaction. Reactions in aqueous phase are generally faster than between a dry gas and a dry solid. Soda lime is commonly used in closed-circuit diving rebreathers and in anesthesia systems.[8][9]
The same catalytic effect by the alkali hydroxides (function of the Na2Oeq content of cement) also contributes to the carbonation of portlandite by atmospheric CO2 in concrete although the rate of propagation of the reaction front is there essentially limited by the CO2 diffusion within the concrete matrix less porous.[10]
Analogy with the alkali-silica reaction[edit]
A similar reaction to above, also catalysed by sodium hydroxide, is the alkali-silica reaction, a slow degradation process causing the swelling and the cracking of concrete containing aggregates rich in reactive amorphous silica. In a very similar way, NaOH greatly facilitates the dissolution of the amorphous silica. The produced sodium silicate then reacts with the calcium hydroxide (portlandite) present in the hardened cement paste to form calcium silicate hydrate (abbreviated as C-S-H in the cement chemist notation). This silicification reaction of Ca(OH)2 on its turn continuously releases again sodium hydroxide in solution, maintaining a high pH, and the cycle continues up to the total disappearance of portlandite or reactive silica in the exposed concrete. Without the catalysis of this reaction by sodium or potassium soluble hydroxides the alkali-silica reaction would not proceed or would be limited to a very slow pozzolanic reaction. The alkali silica reaction can be written like the soda lime reaction, by simply substituting CO2 by SiO2 in the reactions mentioned here above as follows:
reaction 1: SiO2 + NaOH → NaHSiO3 silica dissolution by NaOH:
high pHreaction 2: NaHSiO3 + Ca(OH)2 → CaSiO3 + H2O + NaOH C-S-H precipitation
and regeneration of NaOHsum (1+2): SiO2 + Ca(OH)2 → CaSiO3 + H2O global reaction:
Pozzolanic reaction catalysed by NaOH
See also[edit]
- Carbon dioxide scrubber
- Alkali-silica reaction (ASR)
- Soda–lime glass, a type of glass made from silica, soda, lime, and aluminum oxide
Cobalt glass—known as "smalt" when ground as a pigment—is a deep blue coloured glass prepared by including a cobalt compound, typically cobalt oxide or cobalt carbonate, in a glass melt. Cobalt is a very intense colouring agent and very little is required to show a noticeable amount of colour.
Cobalt glass plates are used as an optical filter in flame tests to filter out the undesired strong yellow light emitted by traces of sodium, and expand the ability to see violet and blue hues,[1] similar to didymium glass.
Moderately ground cobalt glass (potassium cobalt silicate)—called "smalt"—has been historically important as a pigment in glassmaking, painting, pottery, for surface decoration of other types of glass and ceramics, and other media.[2][3] The long history of its manufacture and use has been described comprehensively.[4] Cobalt aluminate, also known as "cobalt blue",[5] can be used in a similar way.
Cobalt glass such as Bristol blue glass is appreciated for its attractive colour and is popular with collectors. It is used in the distinctive blue bottles of Harvey's Bristol Cream sherry and Tŷ Nant mineral water.
https://en.wikipedia.org/wiki/Cobalt_glass
Water (chemical formula H2O) is an inorganic, transparent, tasteless, odorless, and nearly colorless chemical substance, which is the main constituent of Earth's hydrosphere and the fluids of all known living organisms (in which it acts as a solvent[1]). It is vital for all known forms of life, even though it provides no calories or organic nutrients. Its chemical formula H2O, indicates that each of its molecules contains one oxygen and two hydrogen atoms, connected by covalent bonds. The hydrogen atoms are attached to the oxygen atom at an angle of 104.45°.[2] "Water" is the name of the liquid state of H2O at standard conditions for temperature and pressure.
A number of natural states of water exist. It forms precipitation in the form of rain and aerosols in the form of fog. Clouds consist of suspended droplets of water and ice, its solid state. When finely divided, crystalline ice may precipitate in the form of snow. The gaseous state of water is steam or water vapor.
Water covers approximately 70.9% of the Earth's surface, mostly in seas and oceans.[3] Small portions of water occur as groundwater (1.7%), in the glaciers and the ice caps of Antarctica and Greenland (1.7%), and in the air as vapor, clouds (consisting of ice and liquid water suspended in air), and precipitation (0.001%).[4][5] Water moves continually through the water cycle of evaporation, transpiration (evapotranspiration), condensation, precipitation, and runoff, usually reaching the sea.
Water plays an important role in the world economy. Approximately 70% of the freshwater used by humans goes to agriculture.[6] Fishing in salt and fresh water bodies is a major source of food for many parts of the world. Much of the long-distance trade of commodities (such as oil, natural gas, and manufactured products) is transported by boats through seas, rivers, lakes, and canals. Large quantities of water, ice, and steam are used for cooling and heating, in industry and homes. Water is an excellent solvent for a wide variety of substances both mineral and organic; as such it is widely used in industrial processes, and in cooking and washing. Water, ice and snow are also central to many sports and other forms of entertainment, such as swimming, pleasure boating, boat racing, surfing, sport fishing, diving, ice skating and skiing.
Etymology
The word water comes from Old English wæter, from Proto-Germanic *watar (source also of Old Saxon watar, Old Frisian wetir, Dutch water, Old High German wazzar, German Wasser, vatn, Gothic 𐍅𐌰𐍄𐍉 (wato), from Proto-Indo-European *wod-or, suffixed form of root *wed- ("water"; "wet").[7] Also cognate, through the Indo-European root, with Greek ύδωρ (ýdor), Russian вода́ (vodá), Irish uisce, and Albanian ujë.
History
Chemical and physical properties
Water (H
2O) is a polar inorganic compound that is at room temperature a tasteless and odorless liquid, nearly colorless with a hint of blue. This simplest hydrogen chalcogenide is by far the most studied chemical compound and is described as the "universal solvent" for its ability to dissolve many substances.[8][9] This allows it to be the "solvent of life":[10] indeed, water as found in nature almost always includes various dissolved substances, and special steps are required to obtain chemically pure water. Water is the only common substance to exist as a solid, liquid, and gas in normal terrestrial conditions.[11]
States

Along with oxidane, water is one of the two official names for the chemical compound H
2O;[12] it is also the liquidphase of H
2O.[13] The other two common states of matter of water are the solid phase, ice, and the gaseous phase, water vapor or steam. The addition or removal of heat can cause phase transitions: freezing (water to ice), melting(ice to water), vaporization (water to vapor), condensation (vapor to water), sublimation (ice to vapor) and deposition(vapor to ice).[14]
Density
Water differs from most liquids in that it becomes less dense as it freezes.[16] In 1 atm pressure, it reaches its maximum density of 1,000 kg/m3 (62.43 lb/cu ft) at 3.98 °C (39.16 °F).[17] The density of ice is 917 kg/m3(57.25 lb/cu ft), an expansion of 9%.[18][19] This expansion can exert enormous pressure, bursting pipes and cracking rocks (see Frost weathering).[20]
In a lake or ocean, water at 4 °C (39.2 °F) sinks to the bottom, and ice forms on the surface, floating on the liquid water. This ice insulates the water below, preventing it from freezing solid. Without this protection, most aquatic organisms would perish during the winter.[21]
Phase transitions
At a pressure of one atmosphere (atm), ice melts or water freezes at 0 °C (32 °F) and water boils or vapor condenses at 100 °C (212 °F). However, even below the boiling point, water can change to vapor at its surface by evaporation (vaporization throughout the liquid is known as boiling). Sublimation and deposition also occur on surfaces.[14] For example, frost is deposited on cold surfaces while snowflakes form by deposition on an aerosol particle or ice nucleus.[22] In the process of freeze-drying, a food is frozen and then stored at low pressure so the ice on its surface sublimates.[23]
The melting and boiling points depend on pressure. A good approximation for the rate of change of the melting temperature with pressure is given by the Clausius–Clapeyron relation:

where and are the molar volumes of the liquid and solid phases, and is the molar latent heat of melting. In most substances, the volume increases when melting occurs, so the melting temperature increases with pressure. However, because ice is less dense than water, the melting temperature decreases.[15] In glaciers, pressure melting can occur under sufficiently thick volumes of ice, resulting in subglacial lakes.[24][25]



The Clausius-Clapeyron relation also applies to the boiling point, but with the liquid/gas transition the vapor phase has a much lower density than the liquid phase, so the boiling point increases with pressure.[26] Water can remain in a liquid state at high temperatures in the deep ocean or underground. For example, temperatures exceed 205 °C (401 °F) in Old Faithful, a geyser in Yellowstone National Park.[27] In hydrothermal vents, the temperature can exceed 400 °C (752 °F).[28]
At sea level, the boiling point of water is 100 °C (212 °F). As atmospheric pressure decreases with altitude, the boiling point decreases by 1 °C every 274 meters. High-altitude cooking takes longer than sea-level cooking. For example, at 1,524 metres (5,000 ft), cooking time must be increased by a fourth to achieve the desired result.[29] (Conversely, a pressure cooker can be used to decrease cooking times by raising the boiling temperature.[30]) In a vacuum, water will boil at room temperature.[31]
Triple and critical points

On a pressure/temperature phase diagram (see figure), there are curves separating solid from vapor, vapor from liquid, and liquid from solid. These meet at a single point called the triple point, where all three phases can coexist. The triple point is at a temperature of 273.16 K (0.01 °C) and a pressure of 611.657 pascals (0.00604 atm);[32] it is the lowest pressure at which liquid water can exist. Until 2019, the triple point was used to define the Kelvin temperature scale.[33][34]
The water/vapor phase curve terminates at 647.096 K (373.946 °C; 705.103 °F) and 22.064 megapascals (3,200.1 psi; 217.75 atm).[35] This is known as the critical point. At higher temperatures and pressures the liquid and vapor phases form a continuous phase called a supercritical fluid. It can be gradually compressed or expanded between gas-like and liquid-like densities, its properties (which are quite different from those of ambient water) are sensitive to density. For example, for suitable pressures and temperatures it can mix freely with nonpolar compounds, including most organic compounds. This makes it useful in a variety of applications including high-temperature electrochemistry and as an ecologically benign solvent or catalyst in chemical reactions involving organic compounds. In Earth's mantle, it acts as a solvent during mineral formation, dissolution and deposition.[36][37]
Phases of ice and water
The normal form of ice on the surface of Earth is Ice Ih, a phase that forms crystals with hexagonal symmetry. Another with cubic crystalline symmetry, Ice Ic, can occur in the upper atmosphere.[38] As the pressure increases, ice forms other crystal structures. As of 2019, 17 have been experimentally confirmed and several more are predicted theoretically.[39] The 18th form of ice, ice XVIII, a face-centred-cubic, superionic ice phase, was discovered when a droplet of water was subject to a shock wave that raised the water’s pressure to millions of atmospheres and its temperature to thousands of degrees, resulting in a structure of rigid oxygen toms in which hydrogen atoms flowed freely.[40][41] When sandwiched between layers of graphene, ice forms a square lattice.[42]
The details of the chemical nature of liquid water are not well understood; some theories suggest that its unusual behaviour is due to the existence of 2 liquid states.[17][43][44][45]
Taste and odor
Pure water is usually described as tasteless and odorless, although humans have specific sensors that can feel the presence of water in their mouths,[46]and frogs are known to be able to smell it.[47] However, water from ordinary sources (including bottled mineral water) usually has many dissolved substances, that may give it varying tastes and odors. Humans and other animals have developed senses that enable them to evaluate the potability of water by avoiding water that is too salty or putrid.[48]
Color and appearance
Pure water is visibly blue due to absorption of light in the region ca. 600 nm – 800 nm.[49] The color can be easily observed in a glass of tap-water placed against a pure white background, in daylight. The principal absorption bands responsible for the color are overtones of the O–H stretching vibrations. The apparent intensity of the color increases with the depth of the water column, following Beer's law. This also applies, for example, with a swimming pool when the light source is sunlight reflected from the pool's white tiles.
In nature, the color may also be modified from blue to green due to the presence of suspended solids or algae.
In industry, near-infrared spectroscopy is used with aqueous solutions as the greater intensity of the lower overtones of water means that glass cuvettes with short path-length may be employed. To observe the fundamental stretching absorption spectrum of water or of an aqueous solution in the region around 3500 cm−1 (2.85 μm)[50] a path length of about 25 μm is needed. Also, the cuvette must be both transparent around 3500 cm−1 and insoluble in water; calcium fluoride is one material that is in common use for the cuvette windows with aqueous solutions.
The Raman-active fundamental vibrations may be observed with, for example, a 1 cm sample cell.
Aquatic plants, algae, and other photosynthetic organisms can live in water up to hundreds of meters deep, because sunlight can reach them. Practically no sunlight reaches the parts of the oceans below 1,000 meters (3,300 ft) of depth.
The refractive index of liquid water (1.333 at 20 °C (68 °F)) is much higher than that of air (1.0), similar to those of alkanes and ethanol, but lower than those of glycerol (1.473), benzene (1.501), carbon disulfide (1.627), and common types of glass (1.4 to 1.6). The refraction index of ice (1.31) is lower than that of liquid water.
Polar molecule

In a water molecule, the hydrogen atoms form a 104.5° angle with the oxygen atom. The hydrogen atoms are close to two corners of a tetrahedron centered on the oxygen. At the other two corners are lone pairs of valence electrons that do not participate in the bonding. In a perfect tetrahedron, the atoms would form a 109.5° angle, but the repulsion between the lone pairs is greater than the repulsion between the hydrogen atoms.[51][52] The O–H bond length is about 0.096 nm.[53]
Other substances have a tetrahedral molecular structure, for example, methane (CH
4) and hydrogen sulfide (H
2S). However, oxygen is more electronegative (holds on to its electrons more tightly) than most other elements, so the oxygen atom retains a negative charge while the hydrogen atoms are positively charged. Along with the bent structure, this gives the molecule an electrical dipole moment and it is classified as a polar molecule.[54]
Water is a good polar solvent, that dissolves many salts and hydrophilic organic molecules such as sugars and simple alcohols such as ethanol. Water also dissolves many gases, such as oxygen and carbon dioxide—the latter giving the fizz of carbonated beverages, sparkling wines and beers. In addition, many substances in living organisms, such as proteins, DNA and polysaccharides, are dissolved in water. The interactions between water and the subunits of these biomacromolecules shape protein folding, DNA base pairing, and other phenomena crucial to life (hydrophobic effect).
Many organic substances (such as fats and oils and alkanes) are hydrophobic, that is, insoluble in water. Many inorganic substances are insoluble too, including most metal oxides, sulfides, and silicates.
Hydrogen bonding

Because of its polarity, a molecule of water in the liquid or solid state can form up to four hydrogen bonds with neighboring molecules. Hydrogen bonds are about ten times as strong as the Van der Waals force that attracts molecules to each other in most liquids. This is the reason why the melting and boiling points of water are much higher than those of other analogous compounds like hydrogen sulfide. They also explain its exceptionally high specific heat capacity (about 4.2 J/g/K), heat of fusion (about 333 J/g), heat of vaporization (2257 J/g), and thermal conductivity (between 0.561 and 0.679 W/m/K). These properties make water more effective at moderating Earth's climate, by storing heat and transporting it between the oceans and the atmosphere. The hydrogen bonds of water are around 23 kJ/mol (compared to a covalent O-H bond at 492 kJ/mol). Of this, it is estimated that 90% is attributable to electrostatics, while the remaining 10% is partially covalent.[55]
These bonds are the cause of water's high surface tension[56] and capillary forces. The capillary action refers to the tendency of water to move up a narrow tube against the force of gravity. This property is relied upon by all vascular plants, such as trees.[57]
Self-ionisation
Water is a weak solution of hydronium hydroxide - there is an equilibrium 2H
2O ⇔ H
3O+
+ OH−
, in combination with solvation of the resulting hydroniumions.
Electrical conductivity and electrolysis
Pure water has a low electrical conductivity, which increases with the dissolution of a small amount of ionic material such as common salt.
Liquid water can be split into the elements hydrogen and oxygen by passing an electric current through it—a process called electrolysis. The decomposition requires more energy input than the heat released by the inverse process (285.8 kJ/mol, or 15.9 MJ/kg).[58]
Mechanical properties
Liquid water can be assumed to be incompressible for most purposes: its compressibility ranges from 4.4 to 5.1×10−10 Pa−1 in ordinary conditions.[59] Even in oceans at 4 km depth, where the pressure is 400 atm, water suffers only a 1.8% decrease in volume.[60]
The viscosity of water is about 10−3 Pa·s or 0.01 poise at 20 °C (68 °F), and the speed of sound in liquid water ranges between 1,400 and 1,540 meters per second (4,600 and 5,100 ft/s) depending on temperature. Sound travels long distances in water with little attenuation, especially at low frequencies (roughly 0.03 dB/km for 1 kHz), a property that is exploited by cetaceans and humans for communication and environment sensing (sonar).[61]
Reactivity
Metallic elements which are more electropositive than hydrogen, particularly the alkali metals and alkaline earth metals such as lithium, sodium, calcium, potassium and cesium displace hydrogen from water, forming hydroxides and releasing hydrogen. At high temperatures, carbon reacts with steam to form carbon monoxide and hydrogen.
Water cycle

The water cycle (known scientifically as the hydrologic cycle) refers to the continuous exchange of water within the hydrosphere, between the atmosphere, soil water, surface water, groundwater, and plants.
Water moves perpetually through each of these regions in the water cycle consisting of the following transfer processes:
- evaporation from oceans and other water bodies into the air and transpiration from land plants and animals into the air.
- precipitation, from water vapor condensing from the air and falling to the earth or ocean.
- runoff from the land usually reaching the sea.
Most water vapors found mostly in the ocean returns to it, but winds carry water vapor over land at the same rate as runoff into the sea, about 47 Tt per year whilst evaporation and transpiration happening in land masses also contribute another 72 Tt per year. Precipitation, at a rate of 119 Tt per year over land, has several forms: most commonly rain, snow, and hail, with some contribution from fog and dew.[62] Dew is small drops of water that are condensed when a high density of water vapor meets a cool surface. Dew usually forms in the morning when the temperature is the lowest, just before sunrise and when the temperature of the earth's surface starts to increase.[63] Condensed water in the air may also refract sunlight to produce rainbows.
Water runoff often collects over watersheds flowing into rivers. A mathematical model used to simulate river or stream flow and calculate water quality parameters is a hydrological transport model. Some water is diverted to irrigation for agriculture. Rivers and seas offer opportunities for travel and commerce. Through erosion, runoff shapes the environment creating river valleys and deltas which provide rich soil and level ground for the establishment of population centers. A flood occurs when an area of land, usually low-lying, is covered with water which occurs when a river overflows its banks or a storm surge happens. On the other hand, drought is an extended period of months or years when a region notes a deficiency in its water supply. This occurs when a region receives consistently below average precipitation either due to its topography or due to its location in terms of latitude.
Water resources
Water occurs as both "stocks" and "flows". Water can be stored as lakes, water vapor, groundwater or aquifers, and ice and snow. Of the total volume of global freshwater, an estimated 69 percent is stored in glaciers and permanent snow cover; 30 percent is in groundwater; and the remaining 1 percent in lakes, rivers, the atmosphere, and biota.[64] The length of time water remains in storage is highly variable: some aquifers consist of water stored over thousands of years but lake volumes may fluctuate on a seasonal basis, decreasing during dry periods and increasing during wet ones. A substantial fraction of the water supply for some regions consists of water extracted from water stored in stocks, and when withdrawals exceed recharge, stocks decrease. By some estimates, as much as 30 percent of total water used for irrigation comes from unsustainable withdrawals of groundwater, causing groundwater depletion.[65]
Sea water and tides
Sea water contains about 3.5% sodium chloride on average, plus smaller amounts of other substances. The physical properties of seawater differ from fresh water in some important respects. It freezes at a lower temperature (about −1.9 °C (28.6 °F)) and its density increases with decreasing temperature to the freezing point, instead of reaching maximum density at a temperature above freezing. The salinity of water in major seas varies from about 0.7% in the Baltic Sea to 4.0% in the Red Sea. (The Dead Sea, known for its ultra-high salinity levels of between 30–40%, is really a salt lake.)
Tides are the cyclic rising and falling of local sea levels caused by the tidal forces of the Moon and the Sun acting on the oceans. Tides cause changes in the depth of the marine and estuarine water bodies and produce oscillating currents known as tidal streams. The changing tide produced at a given location is the result of the changing positions of the Moon and Sun relative to the Earth coupled with the effects of Earth rotation and the local bathymetry. The strip of seashore that is submerged at high tide and exposed at low tide, the intertidal zone, is an important ecological product of ocean tides.


Effects on life

From a biological standpoint, water has many distinct properties that are critical for the proliferation of life. It carries out this role by allowing organic compounds to react in ways that ultimately allow replication. All known forms of life depend on water. Water is vital both as a solvent in which many of the body's solutes dissolve and as an essential part of many metabolic processes within the body. Metabolism is the sum total of anabolism and catabolism. In anabolism, water is removed from molecules (through energy requiring enzymatic chemical reactions) in order to grow larger molecules (e.g., starches, triglycerides, and proteins for storage of fuels and information). In catabolism, water is used to break bonds in order to generate smaller molecules (e.g., glucose, fatty acids, and amino acids to be used for fuels for energy use or other purposes). Without water, these particular metabolic processes could not exist.
Water is fundamental to photosynthesis and respiration. Photosynthetic cells use the sun's energy to split off water's hydrogen from oxygen.[66] Hydrogen is combined with CO2 (absorbed from air or water) to form glucose and release oxygen.[citation needed] All living cells use such fuels and oxidize the hydrogen and carbon to capture the sun's energy and reform water and CO2 in the process (cellular respiration).
Water is also central to acid-base neutrality and enzyme function. An acid, a hydrogen ion (H+, that is, a proton) donor, can be neutralized by a base, a proton acceptor such as a hydroxide ion (OH−) to form water. Water is considered to be neutral, with a pH (the negative log of the hydrogen ion concentration) of 7. Acids have pH values less than 7 while bases have values greater than 7.
Aquatic life forms
Earth surface waters are filled with life. The earliest life forms appeared in water; nearly all fish live exclusively in water, and there are many types of marine mammals, such as dolphins and whales. Some kinds of animals, such as amphibians, spend portions of their lives in water and portions on land. Plants such as kelp and algae grow in the water and are the basis for some underwater ecosystems. Plankton is generally the foundation of the ocean food chain.
Aquatic vertebrates must obtain oxygen to survive, and they do so in various ways. Fish have gills instead of lungs, although some species of fish, such as the lungfish, have both. Marine mammals, such as dolphins, whales, otters, and seals need to surface periodically to breathe air. Some amphibians are able to absorb oxygen through their skin. Invertebrates exhibit a wide range of modifications to survive in poorly oxygenated waters including breathing tubes (see insect and mollusc siphons) and gills (Carcinus). However, as invertebrate life evolved in an aquatic habitat most have little or no specialization for respiration in water.



As a scientific standard
On 7 April 1795, the gram was defined in France to be equal to "the absolute weight of a volume of pure water equal to a cube of one-hundredth of a meter, and at the temperature of melting ice".[80] For practical purposes though, a metallic reference standard was required, one thousand times more massive, the kilogram. Work was therefore commissioned to determine precisely the mass of one liter of water. In spite of the fact that the decreed definition of the gram specified water at 0 °C (32 °F)—a highly reproducible temperature—the scientists chose to redefine the standard and to perform their measurements at the temperature of highest water density, which was measured at the time as 4 °C (39 °F).[81]
The Kelvin temperature scale of the SI system was based on the triple point of water, defined as exactly 273.16 K (0.01 °C; 32.02 °F), but as of May 2019 is based on the Boltzmann constant instead. The scale is an absolute temperature scale with the same increment as the Celsius temperature scale, which was originally defined according to the boiling point (set to 100 °C (212 °F)) and melting point (set to 0 °C (32 °F)) of water.
Natural water consists mainly of the isotopes hydrogen-1 and oxygen-16, but there is also a small quantity of heavier isotopes oxygen-18, oxygen-17, and hydrogen-2 (deuterium). The percentage of the heavier isotopes is very small, but it still affects the properties of water. Water from rivers and lakes tends to contain less heavy isotopes than seawater. Therefore, standard water is defined in the Vienna Standard Mean Ocean Water specification.
https://en.wikipedia.org/wiki/Water#Dihydrogen_monoxide_parody
The mole (symbol: mol) is the base unit of amount of substance in the International System of Units (SI). It is defined as exactly 6.02214076×1023 particles, which may be atoms, molecules, ions, or electrons.[1]
The definition of mole was adopted in November 2018 as one of the seven SI base units,[1] revising the previous definition that specified one mole as the amount of substance in 12 grams of carbon-12 (12C), an isotope of carbon.
The number 6.02214076×1023 (the Avogadro number) was chosen so that the mass of one mole of a chemical compound in grams is numerically equal, for most practical purposes, to the average mass of one molecule of the compound in daltons. Thus, for example, one mole of water (H2O) contains 6.02214076×1023 molecules, whose total mass is about 18.015 grams and the mean mass of one molecule of water is about 18.015 daltons.
The mole is widely used in chemistry as a convenient way to express amounts of reactants and products of chemical reactions. For example, the chemical equation 2H2 + O2 → 2H2O can be interpreted to mean that for each 2 mol dihydrogen (H2) and 1 mol dioxygen (O2) that react, 2 mol of water (H2O) form. The mole may also be used to measure the amount of atoms, ions, electrons, or other entities. The concentration of a solution is commonly expressed by its molarity, defined as the amount of dissolved substance in mole(s) per unit volume of solution, for which the unit typically used is moles per litre (mol/L), commonly abbreviated M.
The term gram-molecule (g mol) was formerly used for "mole of molecules",[2] and gram-atom (g atom) for "mole of atoms". For example, 1 mole of MgBr2is 1 gram-molecule of MgBr2 but 3 gram-atoms of MgBr2.[3][4]
https://en.wikipedia.org/wiki/Mole_(unit)
Carbon-12 (12C) is the more abundant of the two stable isotopes of carbon (carbon-13 being the other), amounting to 98.93% of element carbon on Earth;[1] its abundance is due to the triple-alpha process by which it is created in stars. Carbon-12 is of particular importance in its use as the standard from which atomic masses of all nuclides are measured, thus, its atomic mass is exactly 12 daltons by definition. Carbon-12 is composed of 6 protons, 6 neutrons, and 6 electrons.
History[edit]
Before 1959, both the IUPAP and IUPAC used oxygen to define the mole; the chemists defining the mole as the number of atoms of oxygen which had mass 16 g, the physicists using a similar definition but with the oxygen-16 isotope only. The two organizations agreed in 1959/60 to define the mole as follows.
This was adopted by the CIPM (International Committee for Weights and Measures) in 1967, and in 1971, it was adopted by the 14th CGPM (General Conference on Weights and Measures).
In 1961, the isotope carbon-12 was selected to replace oxygen as the standard relative to which the atomic weights of all the other elements are measured.[2]
In 1980, the CIPM clarified the above definition, defining that the carbon-12 atoms are unbound and in their ground state.
In 2018, IUPAC specified the mole as exactly 6.022 140 76 × 1023 "elementary entities". The number of moles in 12 grams of carbon-12 became a matter of experimental determination.
Hoyle state[edit]

The Hoyle state is an excited, spinless, resonant state of carbon-12. It is produced via the triple-alpha process, and was predicted to exist by Fred Hoyle in 1954.[3] The existence of the 7.7 MeV resonance Hoyle state is essential for the nucleosynthesis of carbon in helium-burning stars, and predicts an amount of carbon production in a stellar environment which matches observations. The existence of the Hoyle state has been confirmed experimentally, but its precise properties are still being investigated.[4]
The Hoyle state is populated when a helium-4 nucleus fuses with a beryllium-8 nucleus in a high-temperature (108K) environment with densely concentrated (105 g/cm3) helium. This process must occur within 10−16 seconds as a consequence of the short half-life of 8Be. The Hoyle state also is a short-lived resonance with a half-life of 2.4×10−16 seconds; it primarily decays back into its three constituent alpha particles, though 0.0413% of decays (or 1 in 2,421.3) occur by internal conversion into the ground state of 12C.[5]
In 2011, an ab initio calculation of the low-lying states of carbon-12 found (in addition to the ground and excited spin-2 state) a resonance with all of the properties of the Hoyle state.[6][7]
Isotopic purification[edit]
The isotopes of carbon can be separated in the form of carbon dioxide gas by cascaded chemical exchange reactions with amine carbamate.[8]
See also[edit]
https://en.wikipedia.org/wiki/Carbon-12
An isotopical pure diamond is a type of diamond that is composed entirely of one isotope of carbon. Isotopically pure diamonds have been manufacturedfrom either the more common carbon isotope with mass number 12 (abbreviated as 12C) or the less common 13C isotope. Compared to natural diamonds that are composed of a mixture of 12C and 13C isotopes, isotopically pure diamonds possess improved characteristics such as increased thermal conductivity.[1] Thermal conductivity of diamonds is at a minimum when 12C and 13C are in a ratio of 1:1 and reaches a maximum when the composition is 100% 12C or 100% 13C.[1]
Manufacture[edit]
The isotopes of carbon can be separated in the form of carbon dioxide gas by cascaded chemical exchange reactions with amine carbamate.[2] Such CO2can be converted to methane and from there to isotopically pure synthetic diamonds.[3] Isotopically enriched diamonds have been synthesized by application of chemical vapor deposition followed by high pressure.[1]
Types[edit]
Carbon 12[edit]
The 12C isotopically pure, (or in practice 15-fold enrichment of isotopic number, 12 over 13 for carbon) diamond gives a 50% higher thermal conductivitythan the already high value of 900-2000 W/(m·K) for a normal diamond, which contains the natural isotopic mixture of 98.9% 12C and 1.1% 13C. This is useful for heat sinks for the semiconductor industry.[4]
Carbon 13[edit]
Isotopically pure 13C diamond layers 20 micrometers thick are used as stress sensors due to the advantageous Raman spectroscopy properties of 13C.[5]
https://en.wikipedia.org/wiki/Isotopically_pure_diamond
https://en.wikipedia.org/wiki/Internal_conversion
No comments:
Post a Comment